

People with Down syndrome (DS) have an extra copy of chromosome 21, which is the location for the APP gene. This gene codes for a protein called amyloid precursor protein. The leading theory of the cause for Alzheimer's disease (AD) is that plaques in the brain made from amyloid protein causes the symptoms of cognitive decline that define AD. The science behind this theory is not settled, despite the push for drug development to address amyloid protein deposition in the brain as the only means to address Alzheimer's disease.
Morris, Clark and Visser, researchers in Australia, write about the importance of gaining a better understanding for the cause of AD before more drugs are developed. (Morris 2014) They explain further why the amyloid hypothesis has failed clinically based in the failure rate of the five anti-amyloid drugs that have been approved by the FDA. Other theories for the cause of AD include (Armstrong 2013):
- Exacerbation of aging
- Degeneration of anatomical pathways
- Exposure to aluminium
- APOE gene
- Mitochondria dysfunction
- A compromised blood brain barrier
- Immune system dysfunction
- Infectious agents
Armstrong further explained that AD is multifactorial, which is restated by many other authors:
- Alzheimer Disease, a Multifactorial Disorder Seeking Multi-therapies - Igbal 2010
- The multifactorial nature of Alzheimer's disease for developing potential therapeutics.- Carreiras 2013
- The Complex and Multifactorial Nature of Alzheimer’s Disease -Alkadhi 2010
- Toward a multifactorial model of Alzheimer disease - Storandt 2012
Amyloid plaque is estimated to occur in 100% of those with DS by the age of 40, some even younger. However, not all of them experience the dementia symptoms associated with AD. (National Institute of Aging) That fact alone should make it clear to the research and medical community that more than just the APP gene and amyloid protein is involved in the development of AD. This is an important concept, so I'll restate it. If AD was only caused by amyloid protein and the plaques that are made from it then 100% of those with DS would experience the clinical symptoms of AD. This is not the case. This same phenomenon is also seen in those without DS as stated by researchers in Cambridge, "It has been hypothesized that Aβ accumulation is the primary cause of pathogenesis in AD, yet there is a weak correlation between Aβ plaque density and the severity of dementia." (Treusch 2009) More evidence against the amyloid plaque/APP gene theory was just published this month by a team of researchers from the UK and USA. They used a mouse model to triplicate chromosome 21 without the APP gene. These mice still developed amyloid plaques. "Here, we report for the first time that triplication of chromosome 21 genes other than APP, increases amyloid-β aggregation, plaque formation, and cognitive deficits in a novel Down syndrome–Alzheimer’s disease (amyloid-β deposition) mouse model." (Wiseman 2018)
I'll review here factors other than amyloid protein and the APP gene that have been proposed as contributing factors to AD and how those same factors are experienced by people with DS. The management and treatment of these factors, many of which are lifestyle related, can greatly reduce the chances of anyone developing AD, even those with DS. Those factors are:
I'll review here factors other than amyloid protein and the APP gene that have been proposed as contributing factors to AD and how those same factors are experienced by people with DS. The management and treatment of these factors, many of which are lifestyle related, can greatly reduce the chances of anyone developing AD, even those with DS. Those factors are:
- Low T3 (active thyroid hormone)
- Impaired glucose metabolism
- Thiamine deficiency
- Sleep apnea
- Aluminum
- Mitochondria dysfunction
T3 Hormone
The thyroid hormone process within the body is very complicated and involves many steps. A problem in any one of these steps can create a state of hypothyroidism. A key step that is often not recognized as clinically relevant and therefore missed by many physicians is the conversion of T4 (thyroxine) hormone to T3 (triiodothyronine) hormone. This conversion is undertaken by an enzyme called deiodinase that removes an iodine, hence T4 (containing 4 iodine) and it's conversion to T3 (containing 3 iodine). (Image 1) T3 is considered to be "active" thyroid hormone because it is the most important form of thyroid hormone that has cellular activity. T4 is lacking of any major cellular activity. The activity of this deiodinase enzyme is very sensitive to stress, inflammation, nutrient deficiencies, oxidative stress, gastrointestinal issues and several other states of ill-health within the body. (Mancini 2016, Hidal 1988, Vierhapper 1981, DePalo 1994)
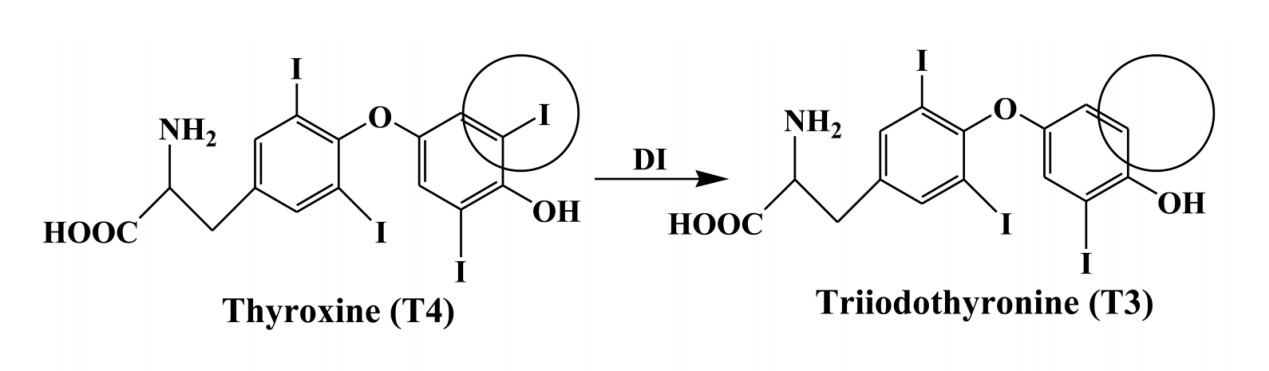
Image 1 Deiodinase 1 converts T4 hormone to T3 hormone.
As it turns out low levels of T3 hormone have been found in those with Alzheimer's. (Karimi 2011) Researchers from Harvard Medical School reviewed this same connection in "Thyroid Function and Alzheimer's Disease". (Tan 2009) The mechanism behind this possible connection is that T3 hormone, as it's considered a transcription factor that effects genetic expression, down-regulates (turns off) the APP gene.(Latasa 1998, Blandia 1998, Belakavadi 2011) This last point should be of great interest to parents, doctors and researchers working with those with DS.
In addition, researchers in Italy found elevated levels of reverse T3 in the cerebral spinal fluid of patients with AD. (Sampaolo 2005). Reverse T3 is a form of T3 hormone created by a different form of deiodinase enzyme that is a sign of aberrant thyroid hormone metabolism. (Biano 2013) Researchers in Japan found that, "Serum rT3 level was a more sensitive parameter than serum T4 or T3 for evaluating thyroid dysfunction." (Shimada 1983) It's also been my experience that reverse T3 levels are elevated in my patients, particularly infants less than one year old as outline here in my research proposal.
In addition, researchers in Italy found elevated levels of reverse T3 in the cerebral spinal fluid of patients with AD. (Sampaolo 2005). Reverse T3 is a form of T3 hormone created by a different form of deiodinase enzyme that is a sign of aberrant thyroid hormone metabolism. (Biano 2013) Researchers in Japan found that, "Serum rT3 level was a more sensitive parameter than serum T4 or T3 for evaluating thyroid dysfunction." (Shimada 1983) It's also been my experience that reverse T3 levels are elevated in my patients, particularly infants less than one year old as outline here in my research proposal.
The role that hypothyroidism plays in the phenotype of DS has been debated ever since the extra chromosome was discovered as the cause of DS in 1958. To this day only one study exists that investigated free T3 and reverse T3 levels in those with DS. They reported no difference in free T3 levels from those in their control group. (Toledo 1997) This has not been my clinical experience. I often see low free T3 levels along with high reverse T3 levels in my younger patients. The majority of my patients I've seen in my practice are aged 3 months - 3 years old. The age range of patients in this study they used was 3 months - 20 years old. That's a significant span in life stages. Thyroid hormone metabolism and the factors that impact it can vary greatly depending on the age of the patient. Much more research is needed that explores the full extent of the thyroid hormone process in those with DS. This research could extend to a better understanding of the role that thyroid hormone plays in the development of AD.
Glucose Metabolism
Of all the organs in the body the brain is the largest consumer of glucose. When at rest, about 60% of the glucose used by the entire body is used by the brain. This is due to it's high rate of metabolism needed for the constant activity including neurotransmitter synthesis, maintaining appropriate electrical charge across the cell membranes and removal of cellular waste and toxins. Many studies exist supporting the connection and likely cause that impaired glucose metabolism has to AD.
Researchers at the Imperial College of London stated, "The impaired glucose metabolism in the brain of subjects with AD is a widely recognized early feature of the disease". (Calsolaro 2016)
- The Alzheimer's disease-related glucose metabolic brain pattern. (Tuene 2014)
- Brain glucose metabolism in Alzheimer's disease. (Swerdlow 1994)
- Sugar and Alzheimer’s disease: a bittersweet truth (Iadecola 2015)
- Abnormalities of glucose metabolism in Alzheimer's disease. (Hoyer 1991)
- Brain fuel metabolism, aging, and Alzheimer's disease. (Cunnane 2011)
- Abnormal Glucose Metabolism in Alzheimer's Disease: Relation to Autophagy/Mitophagy and Therapeutic Approaches. (Banerjee 2015)
- ...and many others
Researchers at the Imperial College of London stated, "The impaired glucose metabolism in the brain of subjects with AD is a widely recognized early feature of the disease". (Calsolaro 2016)
Impaired glucose metabolism is common in children and adults with DS as well. Research corroborates this, especially impaired glucose metabolism in the brain where glucose is needed the most. (Azari 1994, Pietrini 1997, Labudova 1999) Symptoms of this include excessive hunger, excessive thirst, frequent urination, blurred vision, fatigue, frequent infections, tingling in hands and feet. Even the slightest sign of any of these symptoms in those with DS should be taken seriously as they are clues to potentially early stages of glucose metabolism issues. Those who carry extra weight are at even greater risk due to the additional effect this has on insulin resistance.
For a further review of glucose metabolism and the brain I recommend reading "Sugar for the brain: the role of glucose in physiological and pathological brain function".
For a further review of glucose metabolism and the brain I recommend reading "Sugar for the brain: the role of glucose in physiological and pathological brain function".
Thiamine
Thiamine (B1) is heavily involved in the metabolism of glucose. As well, the nervous system is exquisitely sensitive to a thiamine deficiency. Given the recognized connection between impaired glucose metabolism and AD, thiamine has become a likely candidate in the treatment of AD. Gibson, et al have stated, "In animal models, thiamine deficiency exacerbates plaque formation, promotes phosphorylation of tau and impairs memory." (Gibson 2013) In 2010, a more bioavailable form of thiamine, benfotiamine, was given to mice that were genetically altered to create more amyloid protein. They found, "...in the animal Alzheimer's disease model, benfotiamine appears to improve the cognitive function and reduce amyloid deposition via thiamine-independent mechanisms".(Pan 2010) The same research team reported improvement in MMSE (Mini-Mental Status Exam) scores in their subjects with AD who were given benfotiamine. (Pan 2016) They also assessed amyloid deposition and revealed that significant elevations in amyloid deposition occurred despite improvements in cognitive function. This fact reveals that the cognitive deline in AD is due to factors outside of simply plaque deposition. The number of subjects in this study was small but the findings were significant.
The enzyme complex pyruvate dehydrogenase (PDHC) is a key step in glucose metabolism. The cofactors for this enzyme are thiamine (B1), riboflavin (B2) and alpha lipoic acid. PDHC activity was found to be reduced in AD and Huntington's disease indicating a deficiency in one or all of these cofactors. (Sorbi, 1983)
A five year study of 76 elderly patients with mild AD being given benfotiamine is currently underway at Cornell University and will be completed in 2019. (National Institute of Aging) Given the positive results seen in previous studies the results are likely to be positive for some of these patients.
The enzyme complex pyruvate dehydrogenase (PDHC) is a key step in glucose metabolism. The cofactors for this enzyme are thiamine (B1), riboflavin (B2) and alpha lipoic acid. PDHC activity was found to be reduced in AD and Huntington's disease indicating a deficiency in one or all of these cofactors. (Sorbi, 1983)
A five year study of 76 elderly patients with mild AD being given benfotiamine is currently underway at Cornell University and will be completed in 2019. (National Institute of Aging) Given the positive results seen in previous studies the results are likely to be positive for some of these patients.
In addition to it's vital role in glucose metabolism, thiamine is needed for the production of NADPH that is a cofactor the the iodotyrosine deiodinase enzyme (Image 2). This enzyme is found within the thyroid gland itself and scavenges or picks up iodine so it can be recycled to make more thyroid hormone. Low thiamine levels will lead to low NADPH production which will reduce the thyroid glands ability to make thyroid hormone. In addition researchers in Japan found that NADPH also plays a role in T4 to T3 conversion (Sato 1981).
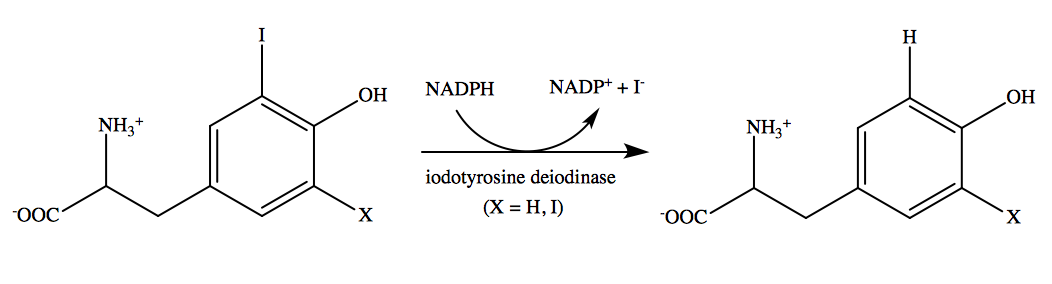
Image 2. NADPH and iodotyrosine deiodinase
To say my experience using high doses of thiamine in the form of benfotiamine in my patients with DS has been interesting is an understatement. While I have not published these findings, I'll report here that many of my young patients and some of my adult patients with DS are experiencing improvements in speech, cognition and gross motor skills at a rate that is much faster than was occurring before benfotiamine was started. I highlight the stories of two patients and explain some of the history and science behind thiamine deficiency in the video linked below if you want to hear more about my experience.
Two published clinical trials exist using thiamine in two different forms in people with DS. The first one was conducted in 1979 and included three adult subjects with DS. While the size of this trial was small the results were significant. Although not clearly stated, the form of thiamine used was likely thiamine HCl or thiamine mononitrate as these are the more common forms used. They gave 50 mg three times a day. The results are recorded in the center column of Table 2 below. (Reading 1979)

Table 2. Results from thiamine supplements to subjects with DS
Despite the significant results from this small trial no further research into the effectiveness or use of thiamine was conducted until 28 years later when another form of thiamine, tetrahydrofurfuryl disulfide (TTFD) was used in a clinical trial. This trial included 22 children with DS between the ages of 8 and 16 years old. They, too, used 50 mg three times per day and included a control group that did not receive TTFD for the first 6 months of the experiment. Their conclusion was, "This study did not reveal any dramatic response in any of our 22 subjects. Although 5 of them did demonstrate some increase in IQ ratings, there was little change in the behavior to encourage the parents in most cases". (Lonsdale 2007) It's possible that TTFD simply isn't the ideal form needed for those with DS.
Again, my experience with using thiamine is in much younger children, and with benfotiamine as the form. To this day a clinical trial investigating the use of benfotiamine (that is non-toxic) in those with DS has not been conducted. My hope is that it would include young children ages 1-3 years given the sensitive nature of brain development at this age.
Again, my experience with using thiamine is in much younger children, and with benfotiamine as the form. To this day a clinical trial investigating the use of benfotiamine (that is non-toxic) in those with DS has not been conducted. My hope is that it would include young children ages 1-3 years given the sensitive nature of brain development at this age.
Sleep apnea
There's no denying the negative effects that hypoxia (low oxygen) has on the brain. Because the brain uses so much glucose it also has a high demand for oxygen. Glucose cannot be optimally used for energy without a sufficient supply of oxygen to match.
Our bodies generate energy from glucose in two ways, aerobic respiration (with oxygen) and anaerobic respiration (without oxygen). Aerobic respiration is the preferred form of energy production because it involves the mitochondria and makes a significantly higher amount of energy. In the absence of oxygen pyruvate (made from glucose) cannot enter the mitochondria. Pyruvate is then converted to lactate and mitochondria sit unused. You can watch the video below to learn more about anaerobic and aerobic respiration.
Our bodies generate energy from glucose in two ways, aerobic respiration (with oxygen) and anaerobic respiration (without oxygen). Aerobic respiration is the preferred form of energy production because it involves the mitochondria and makes a significantly higher amount of energy. In the absence of oxygen pyruvate (made from glucose) cannot enter the mitochondria. Pyruvate is then converted to lactate and mitochondria sit unused. You can watch the video below to learn more about anaerobic and aerobic respiration.
So, in the absence of oxygen the brain can experience elevated levels of lactate, also known as lactic acid. This is the same lactic acid that creates sore muscles after a strenuous workout. A person can feel like they've had a hard workout just from experiencing hypoxia due to obstructive sleep apnea (OSA) at night without the benefits of the strenuous workout. Anyone who as experience with sleep apnea has an increased chance of also experiencing muscle pain. Researchers in Turkey found a "55.4% prevalence of chronic widespread pain in patients with obstructive sleep apnea". (Aytekin 2015) This pain is often due to a build up of lactic acid within muscles.
This same lactic acid can build up in the brain as well and is very toxic to brain cells due to it's acidic nature. In fact, research exists linking elevated lactic acid to amyloid plaques and AD. Researchers at the University of Florida found elevated levels of lactic acid in cerebral spinal fluid of patients with AD. (Xiang 2010) A team in Italy found the same thing in 2015. (Liguori 2015)
The level of hypoxia experienced during OSA varies from patient to patient. Researchers in India measured serum lactate and uric acid levels in patients wit OSA and their conclusion reads, "Both serum UA and lactate were positively correlated with the degree of hypoxia in OSAS. The plasma UA levels in patients with OSAS did not show an overnight rise. However, the plasma lactate levels were higher in the morning. The measurement of serum lactate level was a better marker of oxidative stress among patients with OSAS." (Hira 2012) Earlier in 2009 a research team in Turkey found elevated arterial lactate levels in patients with sleep-related breathing disorders. (Ucar 2009)
The rate of sleep apnea, both obstructive and central, is extremely high in those with DS. Many go undetected and untreated because they experience silent sleep apnea. This form of sleep apnea has no symptoms unless a sleep study is done. The rate of sleep apnea in those with DS has been shown to be as high as 78%. (Fan 2017)
My experience of running more than 500 organic acid tests on children with DS has revealed many things. One of the most significant things it has revealed is the high prevalence of elevated lactic acid in these children, especially with those who are struggling the most in areas of gross motor skill and speech development. An organic acid test is conducted on a first morning urine sample in order to get a more concentrated urine sampe that is the furthest from food consumption that can interfere with results. A first morning urine sample also assesses changes in physiology that occur during sleep.
An elevation in lactic acid can indicate several things: thiamine deficiency, riboflavin deficiency, hypoxia from sleep apnea or other causes of impaired glucose metabolism. I most often see improvements in lactic acid levels after high dose benfotiamine has been started and the test is repeated. I've included three images of lactic acid results from my patients with DS below as examples, but have seen this many more times.
An elevation in lactic acid can indicate several things: thiamine deficiency, riboflavin deficiency, hypoxia from sleep apnea or other causes of impaired glucose metabolism. I most often see improvements in lactic acid levels after high dose benfotiamine has been started and the test is repeated. I've included three images of lactic acid results from my patients with DS below as examples, but have seen this many more times.
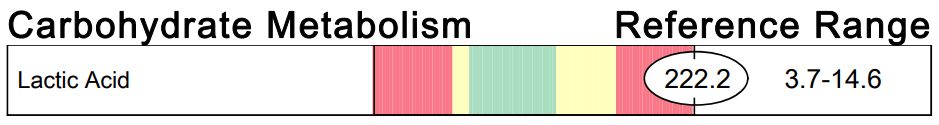
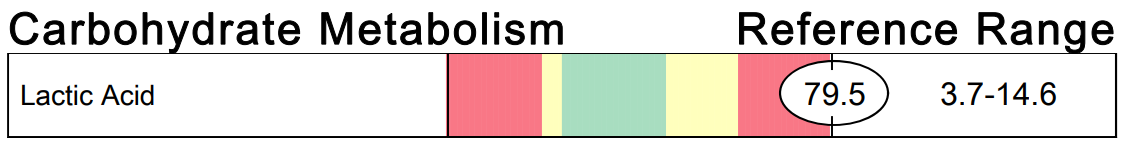
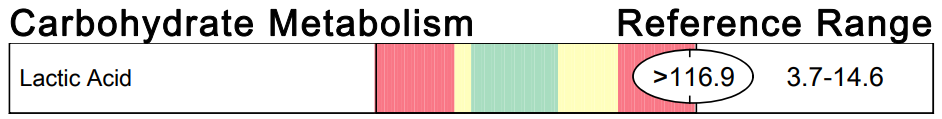
Image 4. Examples of elevated lactic acid in children with Down syndrome
Aluminum
This is one of the oldest and most widely studied theories as the cause of AD. As with all other causes of AD it's also controversial despite the research to support it. The leading world expert on aluminum research is undoubtedly Dr. Chistopher Exley. He is a PhD in ecotoxiclogy of aluminum. He only studies aluminum. He has stated, "Quite simply, all biologically available aluminium in the brain is neurotoxic". (Exley 2016) Researchers in Germany agree with him, writing "Aluminum’s neurotoxic effects in humans and its embryotoxic effects in animal models have been proven." (Klotz 2017) Many others in the scientific community agree and aluminum is now widely accepted to be a neurotoxin.
The main route of aluminum exposure in the body is through digestion as aluminum is the most abundant metal in the earth's crust. Aluminum present in soil eventually makes its way into the food and water we ingest. Fortunately, the lining of our digestive tract creates an effective barrier to the absorption of most consumed aluminum. Unfortunately, this barrier can be disrupted in states of ill-health and increased gastrointestinal permeability, aka leaky gut. Studies have shown that increased aluminum absorption through the GI tract occurs in those with AD.(Moore 2000) Citrate is another factor that can increase aluminum absorption.(Taylor 1998) I often recommend avoiding supplements like magnesium citrate to my patients for this reason.
A secondary source of aluminum that bypasses the gastrointestinal barrier is aluminum hydroxide that is the most common adjuvant used in vaccinations. Exposing the body to aluminum in this way completely bypasses the body's natural means of eliminating it. Aluminum, even in low levels, is a well-established neurotoxin. (Banks 1989, Joshi 1990, Krishnan 1988) As well, there is evidence for aluminum retention within the brain increasing the risk of neurotoxicity with multiple exposures. (Kumar 2014, Gherardi 2015) Dr. Exley and his team tested several commercially available aluminum based adjuvants, including Alhydrogel®, the most commonly used aluminum based adjuvant. This aluminum based adjuvant is in the form of aluminum oxyhydroxide. They revealed that this adjuvant "is most pre-disposed to migration away from the injection site..." to other areas of the body including the brain. (Exley 2016)
There is a growing body of evidence implicating mitochondria dysfuntion as the means by which aluminum causes neuronal damage. (Marchi 2004, Murakami 2004, Niu 2005) It has specifically been shown to interfere with isocitrate dehydrogenase (IDH) that is a NADPH dependent enzyme within the citric acid cycle (Image 5).(Murakami 2004) A decrease in this enzyme makes cells more sensitive to lipid peroxidation and oxidative mitochondria DNA damage. (Kim 2003, Lee 2002)
Aluminum can be a contributing factor to AD for those with DS. It's been shown that those with DS have an increase in gastrointestinal absorption of aluminum. (Moore 1997) It's likely due to the higher incidence and under-recognized rate of SIBO (small intestinal bacterial overgrowth) and gastrointestinal candida overgrowth. (Riordan 2002, Kumamoto 2001)
The main route of aluminum exposure in the body is through digestion as aluminum is the most abundant metal in the earth's crust. Aluminum present in soil eventually makes its way into the food and water we ingest. Fortunately, the lining of our digestive tract creates an effective barrier to the absorption of most consumed aluminum. Unfortunately, this barrier can be disrupted in states of ill-health and increased gastrointestinal permeability, aka leaky gut. Studies have shown that increased aluminum absorption through the GI tract occurs in those with AD.(Moore 2000) Citrate is another factor that can increase aluminum absorption.(Taylor 1998) I often recommend avoiding supplements like magnesium citrate to my patients for this reason.
A secondary source of aluminum that bypasses the gastrointestinal barrier is aluminum hydroxide that is the most common adjuvant used in vaccinations. Exposing the body to aluminum in this way completely bypasses the body's natural means of eliminating it. Aluminum, even in low levels, is a well-established neurotoxin. (Banks 1989, Joshi 1990, Krishnan 1988) As well, there is evidence for aluminum retention within the brain increasing the risk of neurotoxicity with multiple exposures. (Kumar 2014, Gherardi 2015) Dr. Exley and his team tested several commercially available aluminum based adjuvants, including Alhydrogel®, the most commonly used aluminum based adjuvant. This aluminum based adjuvant is in the form of aluminum oxyhydroxide. They revealed that this adjuvant "is most pre-disposed to migration away from the injection site..." to other areas of the body including the brain. (Exley 2016)
There is a growing body of evidence implicating mitochondria dysfuntion as the means by which aluminum causes neuronal damage. (Marchi 2004, Murakami 2004, Niu 2005) It has specifically been shown to interfere with isocitrate dehydrogenase (IDH) that is a NADPH dependent enzyme within the citric acid cycle (Image 5).(Murakami 2004) A decrease in this enzyme makes cells more sensitive to lipid peroxidation and oxidative mitochondria DNA damage. (Kim 2003, Lee 2002)
Aluminum can be a contributing factor to AD for those with DS. It's been shown that those with DS have an increase in gastrointestinal absorption of aluminum. (Moore 1997) It's likely due to the higher incidence and under-recognized rate of SIBO (small intestinal bacterial overgrowth) and gastrointestinal candida overgrowth. (Riordan 2002, Kumamoto 2001)
Mitochondria Dysfunction
Mitochondria are rod-shaped organelles that can be considered the power generators of the cell, converting oxygen and nutrients into adenosine triphosphate (ATP). ATP is the chemical energy "currency" of the cell that powers the cell's metabolic activities. Recall from above that the brain is the most highly metabolic organ of the body, so it's the organ that is most sensitive to impaired mitochondria function.
The evidence pointing to mitochondria dysfunction in AD is strong and growing as well. I've pointed out several means above by which mitochondria function can be impacted. "Mitochondrial function is deregulated in AD and there is growing interest in understanding how altered mitochondrial function may be targeted to inhibit neurodegeneration." (Onyango 2016)
The most basic fuel that mitochondria need to make energy is oxygen and glucose. The process is similar in some ways to the burning of wood to create energy in the form of heat and light from a flame. Glucose in the wood is in the form of cellulose. Wood (glucose) also requires oxygen to burn. The same is true for our bodies. However, because the creation of energy within our bodies is tightly controlled by enzymes it's a bit more complicated than that. Those enzymes that control mitochondria energy production require B vitamins, particularly B1, B2, B3, B5, B6, B7, B9, B12...well, all of them. For those who are familiar with the citric acid cycle and how energy is generated from it, Image 5 shows how extensively the B vitamins are needed for it to operate. To learn more about mitochondria function you can read "Mitochondria - Why they're important and what they need to function".
The evidence pointing to mitochondria dysfunction in AD is strong and growing as well. I've pointed out several means above by which mitochondria function can be impacted. "Mitochondrial function is deregulated in AD and there is growing interest in understanding how altered mitochondrial function may be targeted to inhibit neurodegeneration." (Onyango 2016)
The most basic fuel that mitochondria need to make energy is oxygen and glucose. The process is similar in some ways to the burning of wood to create energy in the form of heat and light from a flame. Glucose in the wood is in the form of cellulose. Wood (glucose) also requires oxygen to burn. The same is true for our bodies. However, because the creation of energy within our bodies is tightly controlled by enzymes it's a bit more complicated than that. Those enzymes that control mitochondria energy production require B vitamins, particularly B1, B2, B3, B5, B6, B7, B9, B12...well, all of them. For those who are familiar with the citric acid cycle and how energy is generated from it, Image 5 shows how extensively the B vitamins are needed for it to operate. To learn more about mitochondria function you can read "Mitochondria - Why they're important and what they need to function".
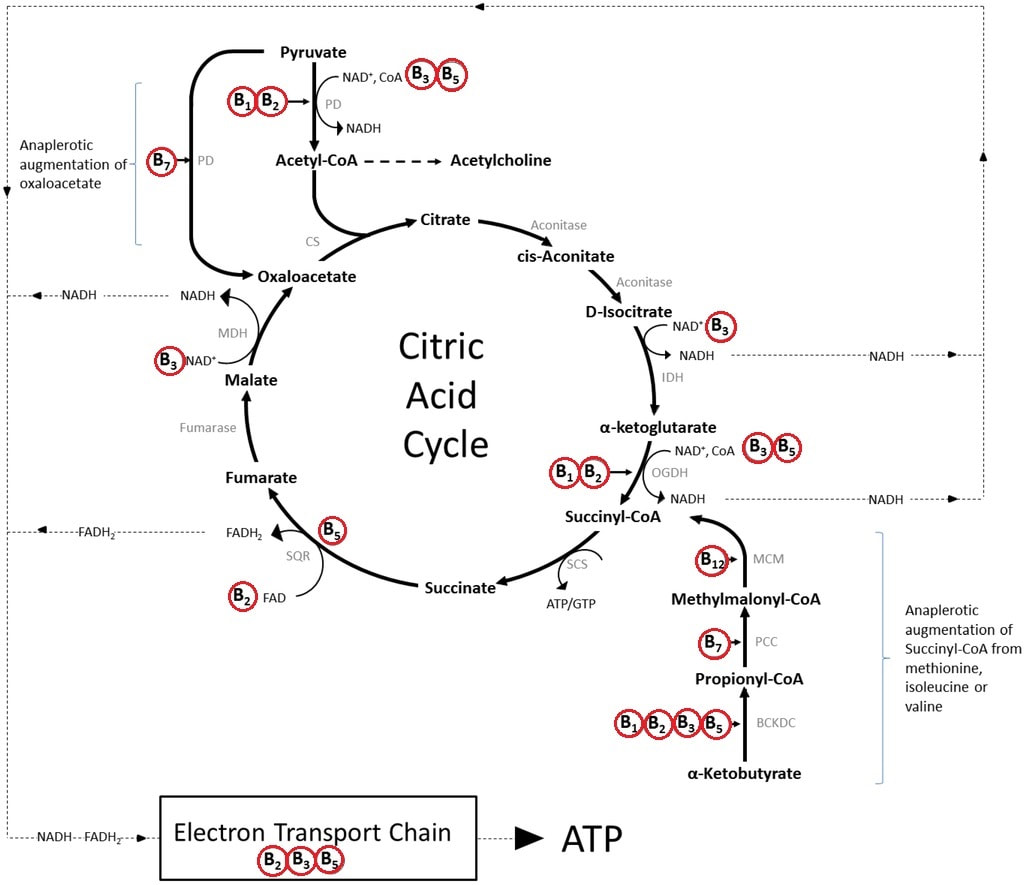
Image 5. B vitamins and the citric acid cycle
It now becomes clear how all of the topics discussed above come together to essentially support mitochondria function. They are truly key players in brain health. Supporting mitochondria function from all angles (sleep, diet, exercise, B vitamins, breathing, avoiding toxic substances, etc) will do a great deal to support brain health and prevent the development of AD in all individuals, including those with DS.
Many studies exist supporting the presence of mitochondria dysfunction in those with DS. Coscun and Busciglio do a thorough job reviewing the role that mitochondria dysfunction has on the phhenotype of those with DS. (Busciglio 2012) In it they state, "Besides oxidative damage, mtDNA mutations and mitochondria dysfunction emerge as important modulators of DS phenotypes." Supporting mitochondria function in those with DS could greatly impact their risk of developing AD.
Many studies exist supporting the presence of mitochondria dysfunction in those with DS. Coscun and Busciglio do a thorough job reviewing the role that mitochondria dysfunction has on the phhenotype of those with DS. (Busciglio 2012) In it they state, "Besides oxidative damage, mtDNA mutations and mitochondria dysfunction emerge as important modulators of DS phenotypes." Supporting mitochondria function in those with DS could greatly impact their risk of developing AD.
Ethical Dilemma
People with DS are being used as guinea pigs to test the safety and efficacy of a new vaccine, ACI-24, designed to stimulate the immune system in a way that it will then destroy amyloid plaques within the brain.(ClinicalTrials.gov) It sounds good on the surface. But when you break it down, they're testing the safety of a previously unused vaccine on people who have a strong history of discrimination and marginalization in order for those without DS to benefit, not to mention the profits that will be made from the creation of such a vaccine. They're testing the safety. This means they don't know if it's safe in humans. Mice studies have been conducted to test the safety and efficacy of ACI-24. (Muse 2007) They tested for signs of inflammation as an indicator of safety. They found no significant elevations in the markers of inflammation. It's unclear how much time lapsed between the end of the injections series and testing for inflammation.
While ACI-24 may indeed create an immune response in the body that attacks amyloid plaque. Is it addressing the real underlying cause of the plaque? What are the consequences to the body if the underlying cause isn't addressed? Let's take thiamine deficiency for example. Thiamine deficiency in a mouse model has been shown to "greatly exacerbate plaque formation" (Gibson 2016) and you now know how thiamine can help prevent and possibly treat AD in humans. So, if we remove the plaque but don't address the thiamine deficiency will the brain and the rest of the body still struggle? It absolutely will. In addition, researchers from Cambridge, MA reviewed the possible protective role that amyloid plaque plays in protecting the brain in "Amyloid deposits - Protection against toxic protein species?". (Treusch 2009). Clearly, more information is needed about the nature of amyloid plaque before a drug or vaccine designed to remove it is developed.
Informed consent is an important and essential part of every ethical human study. Many parents hold guardianship over their adult children with DS and can sign consent for them to receive the experimental vaccine. As well, many adults with DS are perfectly able to understand the implications of, rate of and importance of preventing AD in DS when signing such a consent. However, are parents and those with DS getting all of the information about the yet undetermined cause of AD? Are they being shown or seeing research that supports alternatives to the amyloid plaque theory? Are they truly informed when they sign a consent? I fear not.
Research on human subjects must pass rigorous ethical standards. Guidelines for informed consent have been established by the Office for the Protection of Research Subjects. A full list of the basic elements for informed consent can be found in "Informed consent: Issues and challenges". (Nijhawan 2013). A key aspect of informed consent that should be highlighted here is "A disclosure of any appropriate alternative procedures or courses of treatment that might be advantageous to the subject". Based on what you've read here so far you can see that alternatives do exist. It's likely that subjects and their guardians or parents are not being told of the research supporting alternative means to prevent AD.
The DS population does hold a lot of information about the etiology of AD. However, it's not due to their genetics alone. The metabolic differences experienced in DS hold many clues as well. Researching the effects of addressing these metabolic differences would go a lot further and be much less costly than developing a vaccine that only addresses amyloid plaque.
While ACI-24 may indeed create an immune response in the body that attacks amyloid plaque. Is it addressing the real underlying cause of the plaque? What are the consequences to the body if the underlying cause isn't addressed? Let's take thiamine deficiency for example. Thiamine deficiency in a mouse model has been shown to "greatly exacerbate plaque formation" (Gibson 2016) and you now know how thiamine can help prevent and possibly treat AD in humans. So, if we remove the plaque but don't address the thiamine deficiency will the brain and the rest of the body still struggle? It absolutely will. In addition, researchers from Cambridge, MA reviewed the possible protective role that amyloid plaque plays in protecting the brain in "Amyloid deposits - Protection against toxic protein species?". (Treusch 2009). Clearly, more information is needed about the nature of amyloid plaque before a drug or vaccine designed to remove it is developed.
Informed consent is an important and essential part of every ethical human study. Many parents hold guardianship over their adult children with DS and can sign consent for them to receive the experimental vaccine. As well, many adults with DS are perfectly able to understand the implications of, rate of and importance of preventing AD in DS when signing such a consent. However, are parents and those with DS getting all of the information about the yet undetermined cause of AD? Are they being shown or seeing research that supports alternatives to the amyloid plaque theory? Are they truly informed when they sign a consent? I fear not.
Research on human subjects must pass rigorous ethical standards. Guidelines for informed consent have been established by the Office for the Protection of Research Subjects. A full list of the basic elements for informed consent can be found in "Informed consent: Issues and challenges". (Nijhawan 2013). A key aspect of informed consent that should be highlighted here is "A disclosure of any appropriate alternative procedures or courses of treatment that might be advantageous to the subject". Based on what you've read here so far you can see that alternatives do exist. It's likely that subjects and their guardians or parents are not being told of the research supporting alternative means to prevent AD.
The DS population does hold a lot of information about the etiology of AD. However, it's not due to their genetics alone. The metabolic differences experienced in DS hold many clues as well. Researching the effects of addressing these metabolic differences would go a lot further and be much less costly than developing a vaccine that only addresses amyloid plaque.
Optimal Treatment
A disease with a multifactorial cause requires a multifactorial approach. The work of Dr. Dale Bredesen is the epitome of how this works. It's not likely that I'll be able to fully explain the importance and full breadth of the work being done by Dr. Bredesen. He first published his findings using a multifactorial approach to treating AD in 2014 in his ground-breaking report "Reversal of cognitive decline: A novel therapeutic program". (Bredesen 2014) I was very excited to see that the work he was doing reflected almost exactly what I had been doing to help my patients with DS. It was a very Naturopathic approach that addressed diet, lifestyle, sleep, breathing and supplements. Table 1 below is taken directly from his report and outlines the program he used. While this may seem like a lot at first, it's all actually part of a very healthy lifestyle. There's also a lot of cross-over that occurs between each of these items.
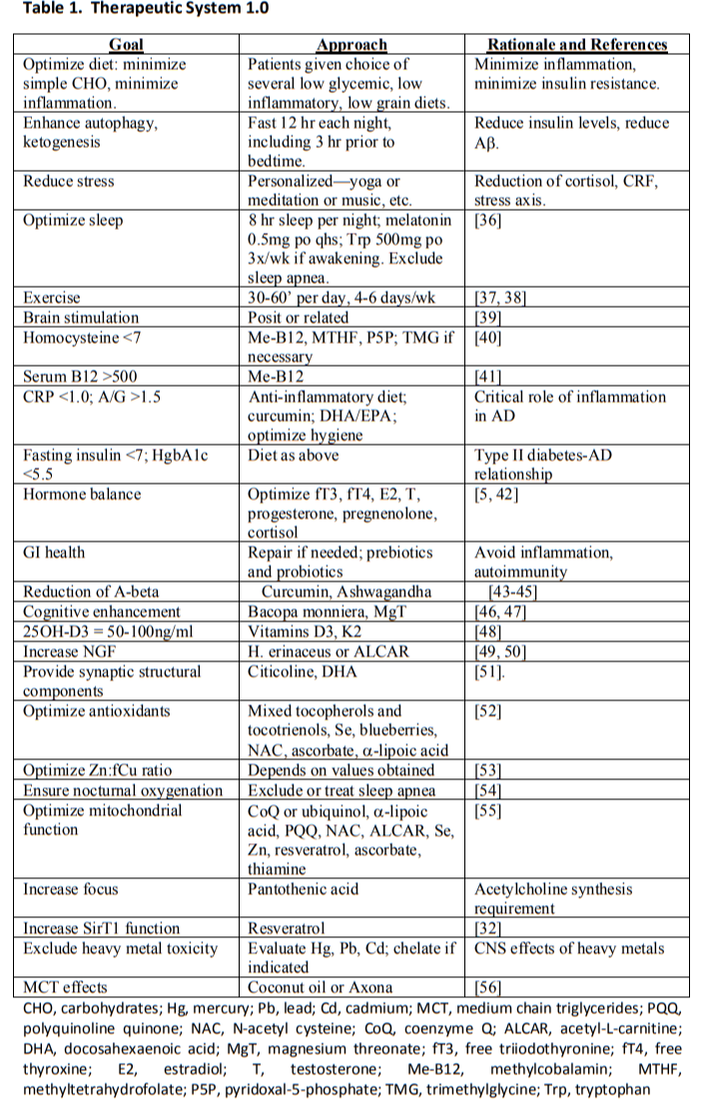
Image 5. Therapeutic system used to reverse cognitive decline (Bredesen 2014)
Dr. Bredesen went on to publish The End of Alzheimer's in 2017. I cannot recommend reading this book more highly. It explains the concepts behind his work in terms that can be understood by lay people as well as appreciated by the medical community. He includes information about the very important APOE gene that has been linked to the increase rate of AD in those with DS as well. "When ApoE4 (ε4) is present in DS, the risk for AD is even higher." (Castro2017) If you're looking to prevent AD in your child or loved one with DS you'll miss a great opportunity to help if you simply wait for a vaccine to become available.
Conclusion
Parents are being misled when they're made to believe that the only way to prevent AD in their child or loved one with DS is to target amyloid plaques that are generated from the APP gene. This only serves to perpetuate the idea that those with an extra copy of chromosome 21 are somehow genetically broken and little can be done to help them other than impacting their defective genetics. Many reading this already know that I wholeheartedly don't believe that people with DS are inherently broken based on their genetic make up. What they need in order to prevent AD is the same thing we all need: a healthy diet, prevention of sleep apnea, avoidance of aluminum and optimization of glucose metabolism.
I'll close by modifying an analogy made by Dr. Bredesen in his book that he calls "36 holes in the roof." The problem of neurodegeneration and dementia in AD is analogous to the problem of a house with many holes in the roof. Some holes are larger than others. Ideally, all holes would be fixed in order to keep the house dry. If a few small holes were left we might be able to manage and keep the house fairly dry inside. Let's imagine that the home owner is convinced that one of the larger holes is the only problem. He works diligently to repair only that hole while standing right outside his front door is a team of people willing to help repair the other holes. The home owner chooses to ignore this team of people and focus only on the one hole. This scenario would be frustrating to witness would it not?
I'll close by modifying an analogy made by Dr. Bredesen in his book that he calls "36 holes in the roof." The problem of neurodegeneration and dementia in AD is analogous to the problem of a house with many holes in the roof. Some holes are larger than others. Ideally, all holes would be fixed in order to keep the house dry. If a few small holes were left we might be able to manage and keep the house fairly dry inside. Let's imagine that the home owner is convinced that one of the larger holes is the only problem. He works diligently to repair only that hole while standing right outside his front door is a team of people willing to help repair the other holes. The home owner chooses to ignore this team of people and focus only on the one hole. This scenario would be frustrating to witness would it not?


 RSS Feed
RSS Feed